小儿磷酸酶过少症(hypophosphatasia)是一种少见的常染色体隐性遗传性疾病,少数为显性遗传。多发生于小儿,其特点是血液、肝脏骨骼和肾脏等组织中的碱性磷酸酶活性低下或消失,骨化不全,易骨折和尿液中磷酰乙醇氨(phosphorylthanolamine)排出量增加。本症于1948年由Rathbun详细综述命名,又称Rathbun综合症,临床较少见。
本病多为常染色体隐性遗传性疾病,少数显性遗传,病因不完全清楚。疑似因体内碱性磷酸酶不足或成骨细胞和软骨细胞的功能不良,所产生的软骨基质和骨样组织性能不佳,碱性磷酸酶的形成减少,钙盐不能正常地沉着。
常染色体隐性遗传通常有以下规律:
(1)假如父母正常,孩子受累,那么父母都是杂合子,并且他们的孩子中平均有1/4的人会受累,1/2是杂合子,1/4正常。
(2)受累者和基因型正常的人结婚生出的孩子都将是表型正常的杂合子。
(3)受累者和杂合子结婚生出的孩子平均1/2将受累,1/2是杂合子。
(4)两个受累者结婚生出的孩子都将受累。
(5)男女受累的机会均等。
(6)杂合子表型正常,但属携带者。假如遗传病是由于某一特定蛋白质缺失所致(例如酶),那么携带者一般来说这种蛋白质的量会减少。假如已经知道突变的部位,分子遗传学分析可以鉴定表型正常的杂合子个体。近亲婚配在常染色体隐性遗传病中有重要作用。有血缘关系的人更容易携带同一个突变等位基因。
常染色体显性遗传,是指缺陷基因呈显性表达,50%的子女都有发病可能性的遗传性疾病。通常有以下几条规律:
(1)受累者的父母中有一方受累;
(2)受累者和未受累者所生的孩子中,受累和未受累的平均数相等;
(3)父母中有一方受累而本人未受累时,他的子孙也不会受累;
(4)男女受累的机会相等;
(5)受累者子女中出现病症的发生率为50%。
查看详细>>
根据临床症状出现的早晚分为新生儿型(又称先天性致死型(congenital lethal hypophosphatasia))、婴儿型、儿童型和青少年型(hypophosphatasia tarda)。但上述各型并无截然分界,常常是病情修复和好转过程的延续表现。
新生儿型于出生后即出现颅骨软化、肢体短粗、弯曲畸形及骨折,常伴有肢体表面皮肤的环形深凹切迹,有些病例可见蓝色巩膜(巩膜外观呈均匀的亮蓝色或蓝灰色,而角膜缘外稍显淡白色)、惊厥、青紫等症状,严重者出生时即为死胎,因胸廓软弱无力和呼吸困难易致死亡。
婴儿型常于生后1~6个月发病,表现为体重不增,生长缓慢及头围增长亦缓慢,颅骨软化、颅缝增宽、囟门突出、头皮静脉怒张、胸廓畸形和四肢弯曲。当高钙血症和肾功能损伤时,可出现厌食,呕吐、便秘、发热、多饮多尿,肌张力低下、惊厥,蛋白尿或脓尿。有些患儿可见皮肤色素沉着。
当病变自然好转,延续到2岁以后,可见儿童型症状,如走路较晚、步态不稳、肢体疼痛、弯曲畸形,牙齿发育不良等。龋齿和乳牙早脱亦属常见。因颅缝早闭,常发生继发性颅压增高或神经系统症状。发病年龄越大,临床症状越不典型,病变亦较轻。个别患儿仅表现为乳牙早脱、骨骼病变不明显。
成人型常只感觉到骨疼的症状,容易骨折以及小儿时期病变所遗留的轻重不等的骨畸形。少数病人在关节、肌腱和椎间韧带周围发生钙化。
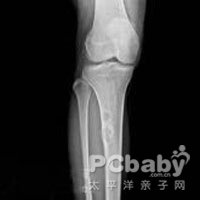
临床表现特点,血、尿生化检查和X线照片,是诊断本症的主要依据。
查看详细>>
根据临床症状出现的早晚分为新生儿型(又称先天性致死型(congenital lethal hypophosphatasia))、婴儿型、儿童型和青少年型(hypophosphatasia tarda)。但上述各型并无截然分界,常常是病情修复和好转过程的延续表现。
新生儿型于出生后即出现颅骨软化、肢体短粗、弯曲畸形及骨折,常伴有肢体表面皮肤的环形深凹切迹,有些病例可见蓝色巩膜(巩膜外观呈均匀的亮蓝色或蓝灰色,而角膜缘外稍显淡白色)、惊厥、青紫等症状,严重者出生时即为死胎,因胸廓软弱无力和呼吸困难易致死亡。
婴儿型常于生后1~6个月发病,表现为体重不增,生长缓慢及头围增长亦缓慢,颅骨软化、颅缝增宽、囟门突出、头皮静脉怒张、胸廓畸形和四肢弯曲。当高钙血症和肾功能损伤时,可出现厌食,呕吐、便秘、发热、多饮多尿,肌张力低下、惊厥,蛋白尿或脓尿。有些患儿可见皮肤色素沉着。
当病变自然好转,延续到2岁以后,可见儿童型症状,如走路较晚、步态不稳、肢体疼痛、弯曲畸形,牙齿发育不良等。龋齿和乳牙早脱亦属常见。因颅缝早闭,常发生继发性颅压增高或神经系统症状。发病年龄越大,临床症状越不典型,病变亦较轻。个别患儿仅表现为乳牙早脱、骨骼病变不明显。
成人型常只感觉到骨疼的症状,容易骨折以及小儿时期病变所遗留的轻重不等的骨畸形。少数病人在关节、肌腱和椎间韧带周围发生钙化。
临床表现特点,血、尿生化检查和X线照片,是诊断本症的主要依据。
查看详细>>
重症病人常于生后不久或1岁内死亡。病情轻者随年龄增长骨病变可自行修复,临床症状可减轻,血、尿生化改变也可有所改善,但仍不正常。
本病除对症治疗外,尚无特效方法。高磷酸盐剂(中性磷酸钠)持续治疗可使血磷轻度升高,增加尿焦磷酸盐的排泄,X线显示骨钙化明显改善。Bongiovanni等报告3例患者用中性磷酸钠1.25~3.0g/d,分4~5次口服,可使本病有改善。
对高钙血症可给低钙饮食或加用可的松治疗。骨骼畸形严重和颅缝早期愈合的病例可考虑采用外科手术。目前已经能应用敲除基因小鼠制作低磷酸酯酶症的动物模型及进行基因研究,基因治疗才能从根本上改变此病的预后。
查看详细>>
预防措施应从孕前贯穿至产前。
婚前体检在预防出生缺陷中起到积极的作用,作用大小取决于检查项目和内容,主要包括血清学检查(如乙肝病毒、梅毒螺旋体、艾滋病病毒)、生殖系统检查(如筛查宫颈炎症)、普通体检(如血压、心电图)以及询问疾病家族史、个人既往病史等,做好遗传病咨询工作。
孕妇尽可能避免危害因素,包括远离烟雾、酒精、药物、辐射、农药、噪音、挥发性有害气体、有毒有害重金属等。在妊娠期产前保健的过程中需要进行系统的出生缺陷筛查,包括定期的超声检查、血清学筛查等,必要时还要进行染色体检查。
一旦出现异常结果,需要明确是否要终止妊娠;胎儿在宫内的安危;出生后是否存在后遗症,是否可治疗,预后如何等等。采取切实可行的诊治措施。
所用产前诊断技术有:
1、羊水细胞培养及有关生化检查(羊膜穿刺时间以妊娠16~20周为宜);
2、孕妇血及羊水甲胎蛋白测定;
3、超声波显像(妊娠4个月左右即可应用);
4、X线检查(妊娠5个月后),对诊断胎儿骨骼畸形有利;
5、绒毛细胞的性染色质测定(受孕40~70天时),预测胎儿性别,以帮助对X连锁遗传病的诊断;
6、应用基因连锁分析;
7、胎儿镜检查。
通过以上技术的应用,防止患有严重遗传病和先天性畸形胎儿的出生。
查看详细>>